What is the 'folding problem' of proteins?
by
It is known that each protein has a unique three-dimensional structure for the proteins that are the core of all biological processes, but research on 'what kind of three-dimensional structure each protein actually has' Hasn't made much progress in the last 50 years. Jason Crawford, who was actually doing research on folding, explains how difficult it is to study the process of ' folding ' leading up to the three-dimensional structure of each protein.
What is “protein folding”? A brief explanation
https://rootsofprogress.org/alphafold-protein-folding-explainer
A protein is a general term for a large molecule in which a large number of 21 types of amino acids are bound, and the number and types of amino acids that make up each protein and the order of binding are determined by the base sequence of DNA. All proteins have a linear shape called a chain, but in reality, each protein gathers and stabilizes in a three-dimensional shape rather than a linear shape.
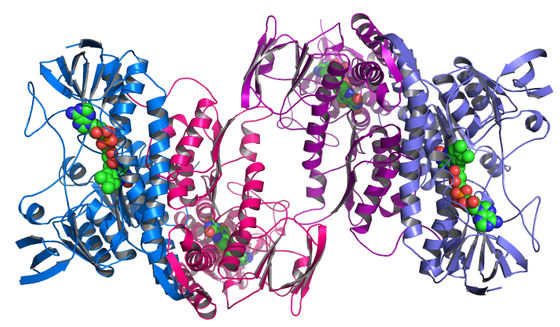
When each protein is considered as a chain, it is called a ' primary structure ', and when it is considered as a three-dimensional shape, it is called a ' secondary structure '. Typical examples of the secondary structure of proteins are the following β-sheets (left figure) and α-helix (right figure). The β-sheet is a stable state in which one protein is folded in a plane, and the α-helix is It refers to a spiral and stable state.
.png)
By
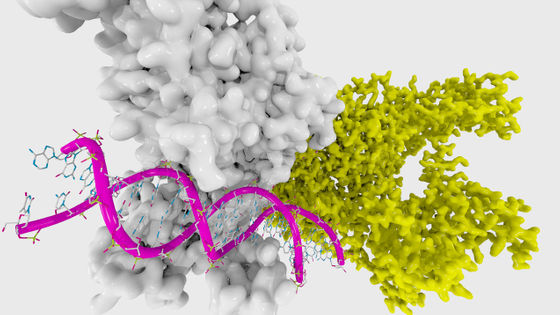
The state in which proteins with secondary structure are gathered and stabilized is called ' tertiary structure '. An example of tertiary structure is the tertiary structure of a bacterial enzyme called Colwellia psychrerythraea.
by Argonne National Laboratory
Tertiary structure looks like a random structure in which multiple proteins are entwined, but in reality there is only one tertiary structure for each protein. Since each protein has properties depending on its tertiary structure, it is important to investigate the tertiary structure, but current methods for investigating tertiary structure are expensive, time-consuming, and cannot be applied to some proteins. It is impossible to investigate all the proteins that have already been found in 180 million. Therefore, there is a need for a method to solve the 'folding problem' of how each protein folds and to estimate the tertiary structure from the primary structure.
Computer simulators are used to solve folding problems. The tertiary structure can be estimated by inputting a model that considers the position, charge, chemical bond, etc. of atoms contained in each protein into the simulator and calculating the acceleration and velocity of each, and this kind of academic field is ' molecular power '. It seems that it has become popular in recent years as a ' learning method '.
However, the method of estimating tertiary structure with a simulator has the problem that it requires the power of a computer. The majority of proteins are composed of thousands of atoms and also interact with surrounding water molecules. Therefore, there are about 30,000 atoms involved in one general tertiary structure, and the mutual relationship reaches 450 million. Instead of simulating all the atoms, an alternative method of calculating the most stable structure from the possible structure candidates from the energy landscape is also devised, but it is expected that the number of structure candidates will reach 10 300. It is said that the life of the universe will expire before all patterns are calculated.
Supercomputers and distributed computing have helped with these calculations. The project ' Folding @ home ', which solves the folding problem by adding up the computing power of PCs in homes around the world, has reached the combined performance of the world's TOP500 supercomputers, and since the pandemic of the new coronavirus after 2020 , We are also analyzing the proteins inside the new coronavirus.
The computing speed of 'Folding @ home', which advances the analysis of the new coronavirus using PCs around the world, reaches the total performance of all the world's TOP500 supercomputers --GIGAZINE

It was a folding problem that was being promoted in this way, but on December 1, 2020, Google's artificial intelligence company DeepMind called ' AlphaFold ' that can predict the tertiary structure of proteins at a dramatic speed and with high accuracy using machine learning. Announced the system.
'Alpha Fold' developed by DeepMind has shown a path with the power of AI and accelerated research to the super difficult problem of biology from 50 years ago --GIGAZINE

AlphaFold learns various functions related to each protein through multiple neural networks and derives a function that can predict the final distance of amino acids contained in proteins in tertiary structure. It has been confirmed that the structure prediction by AlphaFold not only exceeds other computer programs, but also achieves accuracy that exceeds the conventional method.
DeepMind claimed in its announcement that it 'solved the folding problem,' and Crawford commented, 'I think DeepMind's claim is too simple, but it's a breakthrough anyway.' I will.
Related Posts:




in Science, Posted by darkhorse_log